Using pathogen genomics to improve public health.
Genomic data is a useful tool for tracking infectious disease spread, but there are still conceptual and practical barriers to applying pathogen genomics to pressing public health questions. We aim to harness the full potential of sequencing data to understand and combat human pathogens through empirical study and methodological innovation. This involves working towards a more complete understanding of the evolutionary mechanisms of both viral and bacterial disease-causing pathogens, and finding better ways to account for sampling biases in phylogenetic and other analytical results. We are also passionate about ensuring that sequencing capacity and expertise are in the hands of local public health experts who are best positioned to take actionable steps towards responding to pathogen threats.
Cholera Genomics
Sequencing data can improve our understanding of pathogen transmission. This is especially important for pathogens like Vibrio cholerae—the bacteria that causes cholera—that regularly cause thousands of cases and deaths around the globe. Our cholera research focuses on taking a regional approach to understanding transmission in sub-Saharan Africa (e.g., by performing joint phylogenetic analyses of whole genome sequences from multiple countries, as seen in Ekeng et al. 2021, on the right), where Vibrio cholerae causes large outbreaks of often unexplained origin. Additionally, a better understanding of the more complex forms of variation common in bacteria may also lead to insights in both disease pathogenesis and transmission, and we are actively working to improve bioinformatics methods for bacterial assembly and analysis, and to make them easily accessible to public health experts across the globe. Our pipelines—developed in partnership with the CholGen Consortium (funded by the Bill and Melinda Gates Foundation)—are publicly available at https://github.com/cholgen.

Viral Genomics
We also use genomic data to answer questions of pathogen origins, evolution, and transmission in the context of viral outbreaks. During the COVID-19 pandemic, for example, we showed that there had already been several introductions of SARS-CoV-2 into the National Capital Region (DC, Maryland, Virginia) by early March 2020, just a few weeks into official declaration of the pandemic. We also showed that international border closures played a limited role in reducing COVID-19 transmission across the busy US-Mexico border. These independent studies both have important implications for how we think about and enact containment measures during future outbreaks.
We have also more explicitly examined the role of genomic data in supplementing information gathered during “traditional” epidemiological investigations. As shown at right (Wohl et al. 2020), genomic data can both reconstruct known transmission links and also suggest many more connections that may otherwise have been missed. This type of analysis led to the connection of a seemingly university-based mumps virus outbreak in Massachusetts to cases in the broader community.
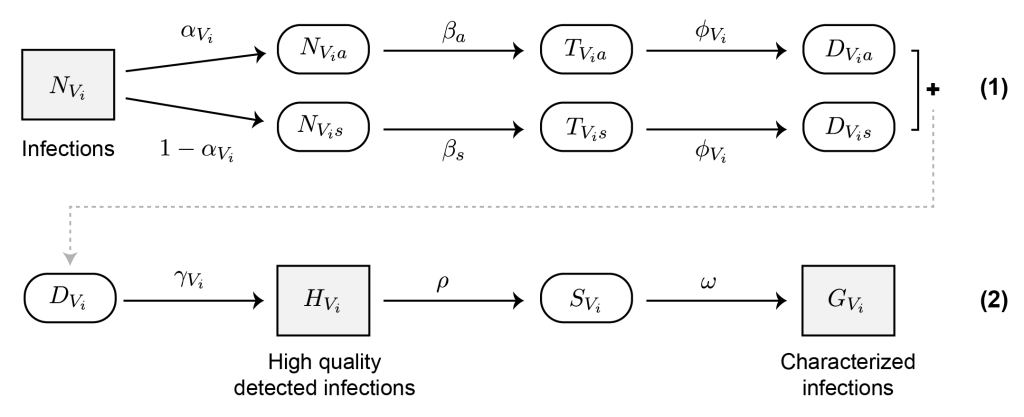
Sampling Theory
Sample size calculations are a crucial piece of using genomics and phylogenetics for public health impact, but there is a lack of clear guidance for determining the sample size needed for phylogenetic and other genomics-based studies. Therefore, our research also includes theoretical and mathematical work aimed at providing a basic framework for these sorts of sampling calculations (e.g., as shown at right, from Wohl et al. 2022). As above, we aim to make our work both publicly available (https://hopkinsidd.github.io/phylosamp/) and easy-to-use by public health professionals, while also remaining flexible enough to be adapted to various pathogens and surveillance scenarios. Current efforts include expanding our existing framework to wastewater sequencing and better integrating epidemiological information into our definitions of phylogenetic linkage.